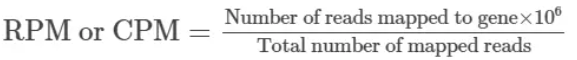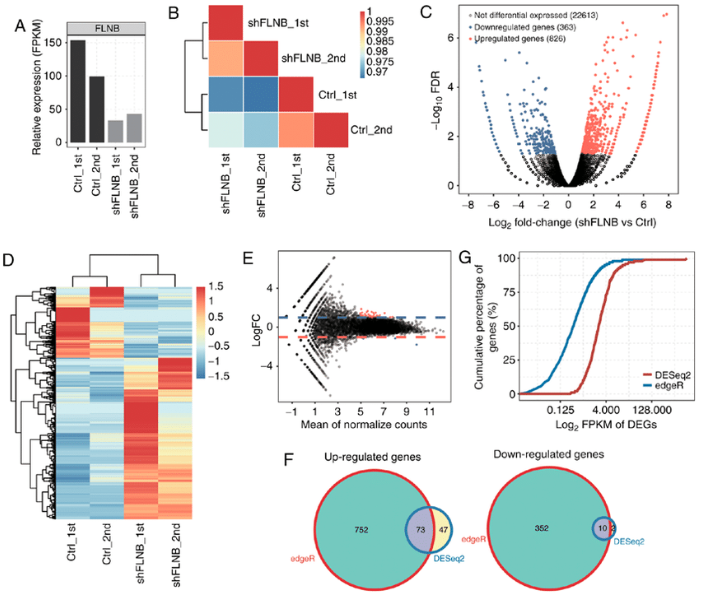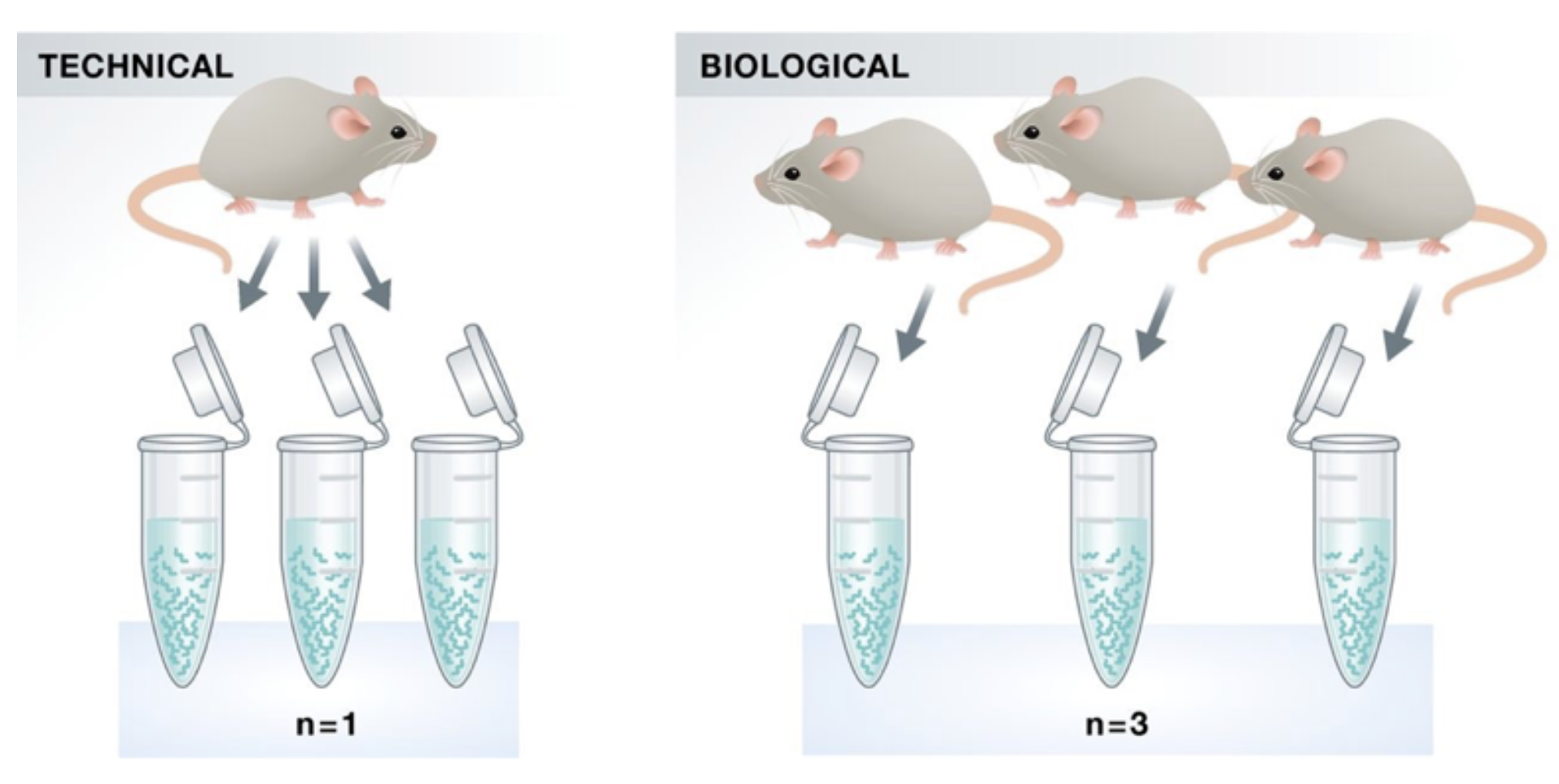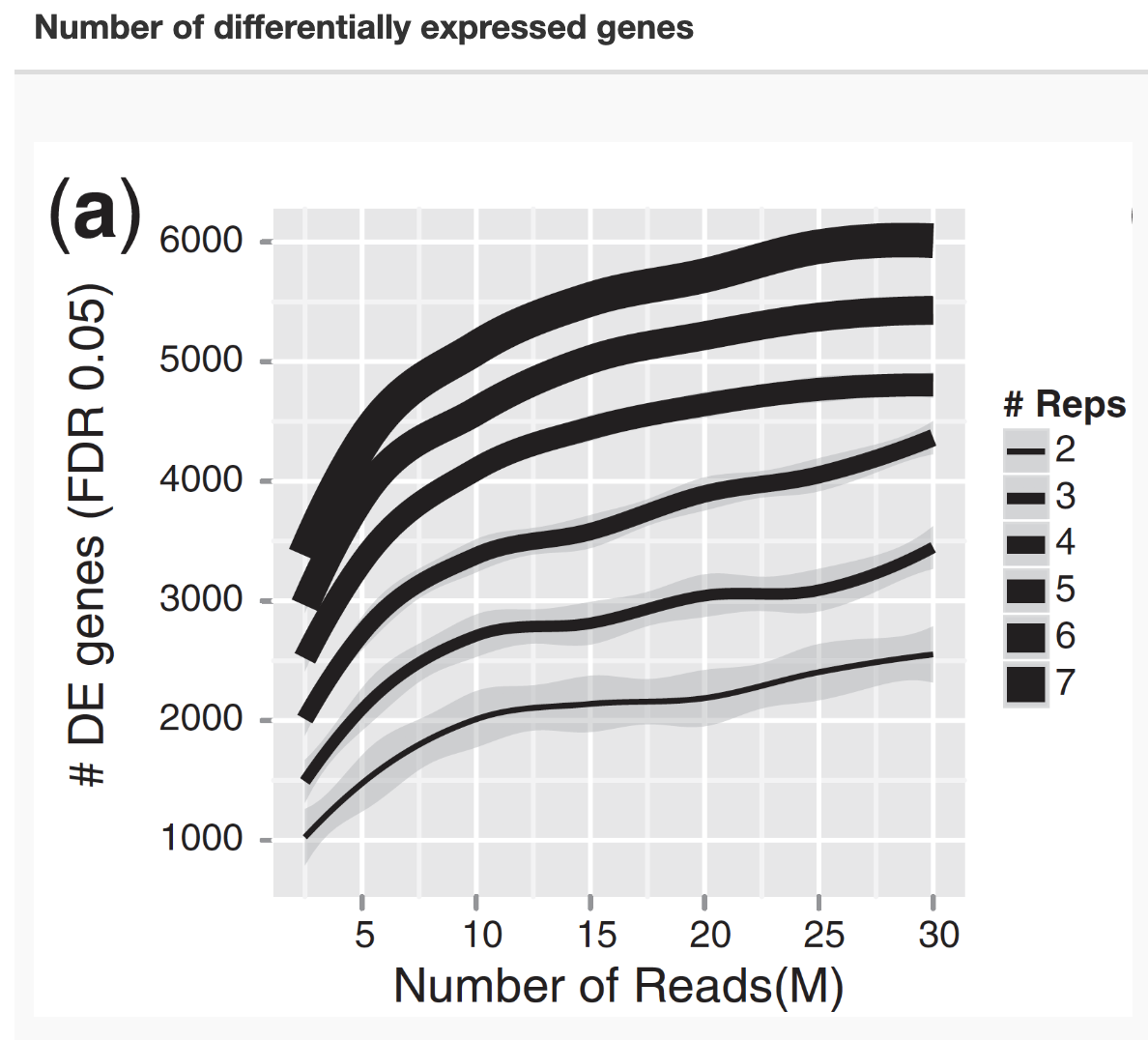

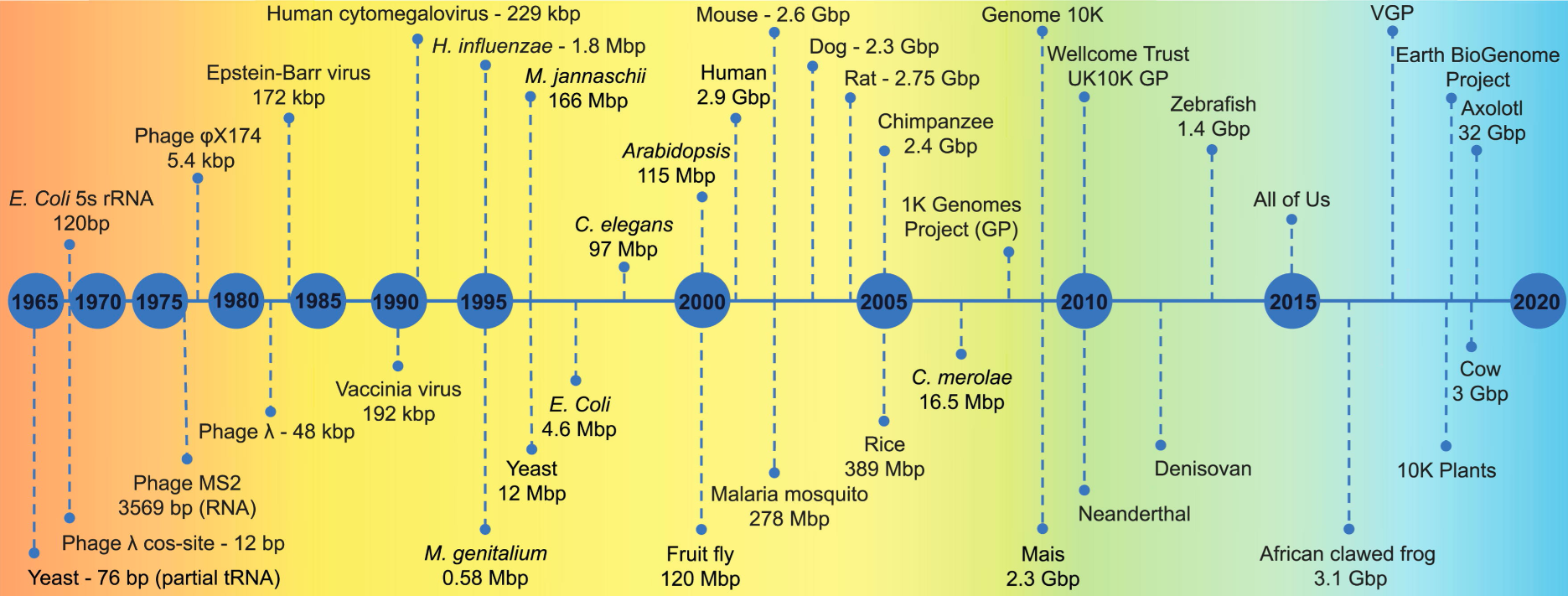


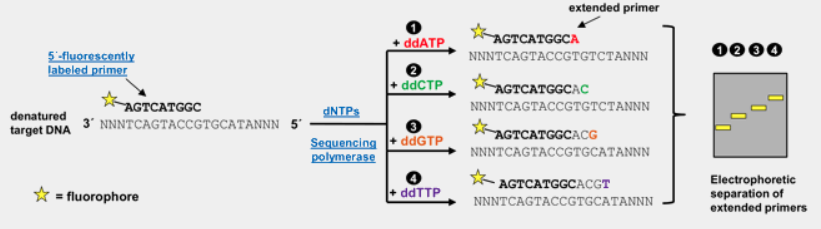
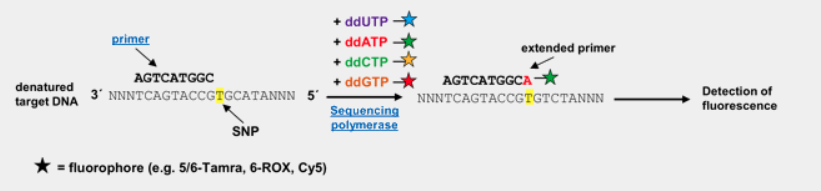
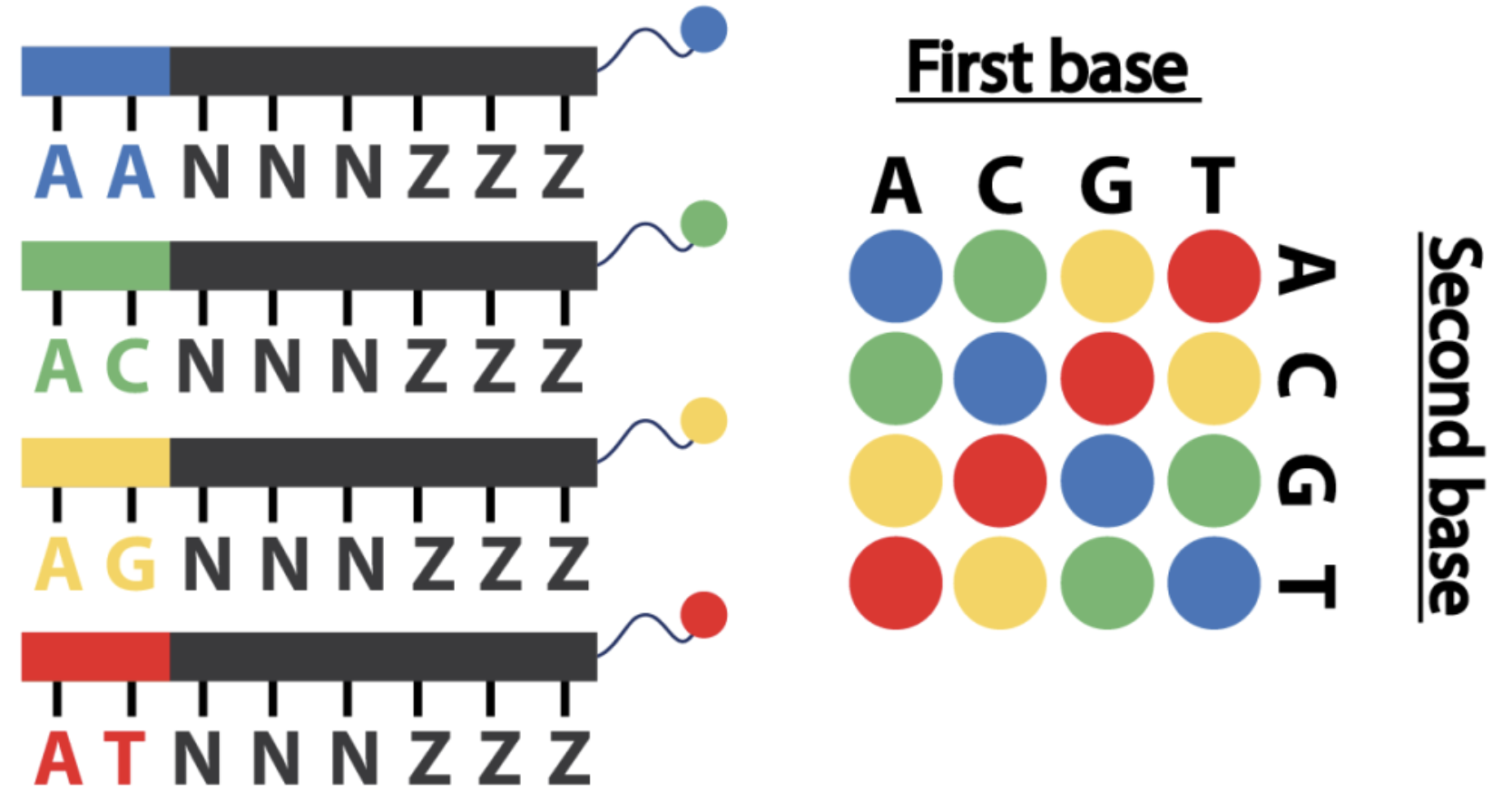



flowchart TB
subgraph vitro [In vitro]
direction TB
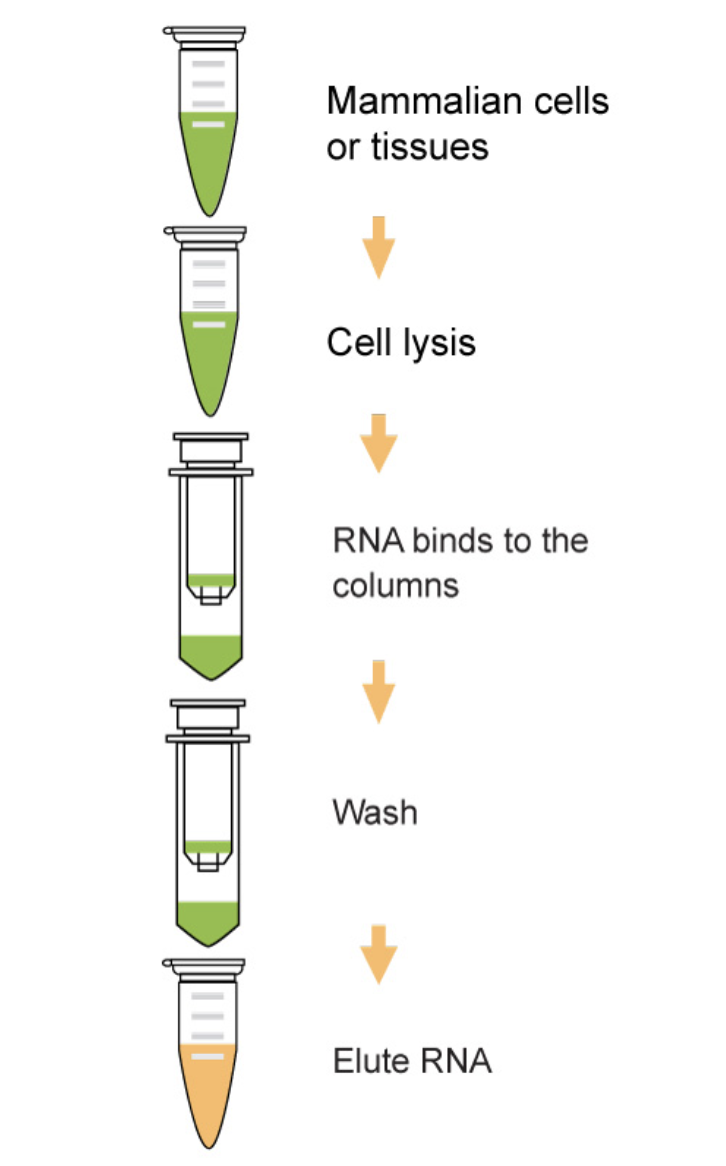
A(<font size=6>Isolate total RNA) --> B(<font size=6>Enrich a specific type of RNA)
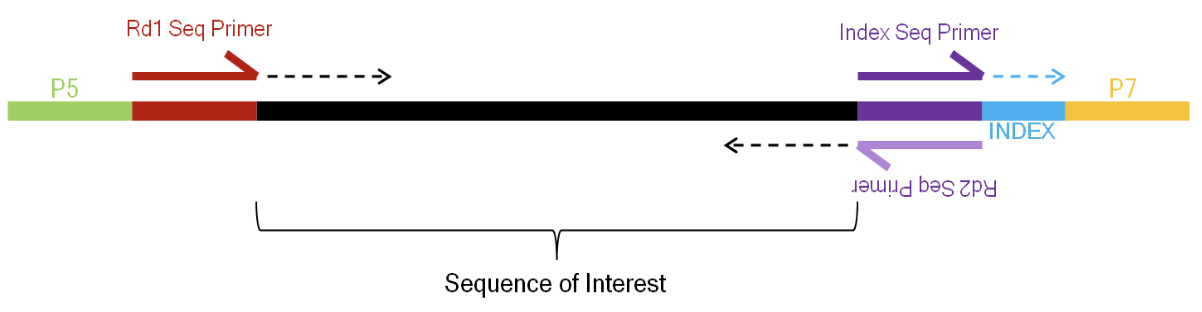
B --> C(<font size=6>Prepare the RNA sequencing library)
end
subgraph silico [In silico]
direction TB
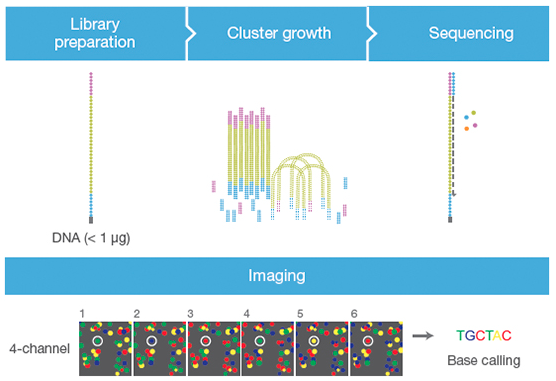
D(<font size=6>Sequencing) --> E(<font size=6>Quantify expression)
E --> F(<font size=6>Differential Expression analysis)
F --> G(<font size=6>Biological conclusions)
end
vitro --> silico
classDef className fill:#D1D1D1,stroke:#333,stroke-width:1px
class A,B,C,D,E,F,G className;